


We have created a awesome theme
Far far away,behind the word mountains, far from the countries
 Medical Device Consulting > Domestic(korea)
Medical Device Consulting > Domestic(korea)
Medical Device Consulting
Domestic(Korea) Consulting
GMP
GMP : Good Manufacturing Practice
▶ GMP?
 |
Medical Device GMP process is a quality assurance system in Korea for securing safety and effectiveness by confirming of manufacturing product as intended. This process is requiring checking the overall quality management process in design, development, and production and after production. In 2011, KGMP for importer’s foreign manufacturer became mandatory. This replaced KGIP (Korea Good Importing Practice). KGMP requirements are based on ISO 13485 |
|---|
Each manufacturing site that intends to market their products in South Korea is required to meet KGMP requirements, submit applicable documents and undergo review. The requirements are described below
- Number of Employees
- List of medical devices manufactured at the facility
- ISO 13485 certificate
- Layout of manufacturing facility
- List of instruments in manufacturing facility
- Major supplier list, outsourced work responsibility/flow
- Legal manufacturer & manufacturing facility relationship evidence document (e.g. FSC, ISO certificate)
- External ISO 13485/GMP audit reports
- Quality manual
- Device master record (Index, Summary or Extracted Content)
- Service manual
▶ Relevant Regulations
·
·
▶ GMP Process
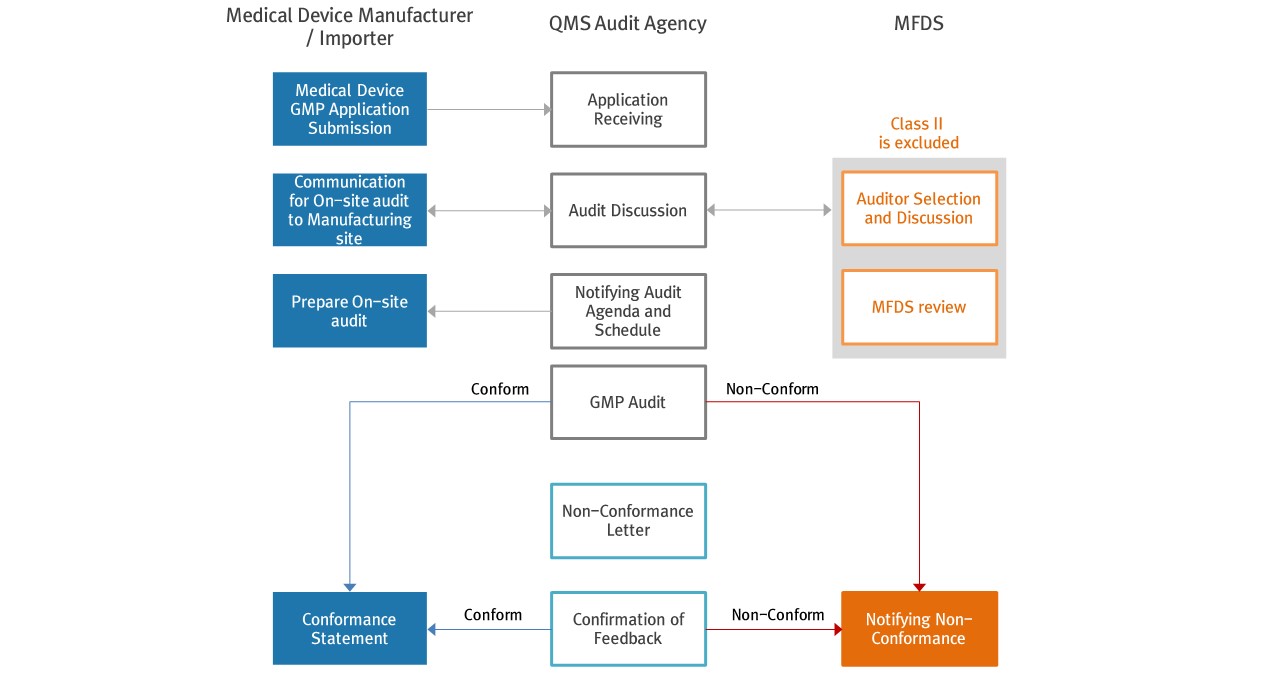
▶ Conformance acceptance categories
- Initial inspection: Initial inspection for newly registered product and manufacture
- Regular inspection: Every 3 years after initial inspection (should apply 90 days before expiration)
- Additional inspection: Adding new product category in scope
- Change inspection: Manufacturing address changes (except small storage area and laboratory which is not related with product quality)

MFDS
MFDS : Ministry of Food and Drug Safety
▶ Applicable Device
According to Korea Medical Device Law Chapter 2, Medical Devices and devices under the definition of Medical Accessories should acquire Manufacturing approval or importing approval for their sales in Korea.
Korea Medical Device Law Chapter 2 (Definition)
Any apparatus, equipment, tool and material products to be used to Human or animal as independently or combined with other device are defined as Medical Device or accessories. Those are any products under below categories (except drug and others in Pharmaceutical law, Devices for Handicapped person)
- Diagnosis, prevention, monitoring, treatment, or alleviation of disease;
- Diagnosis, monitoring, treatment, alleviation, or compensation for an injury or handicap;
- Investigation, replacement, or modification of the anatomy or of a physiological process;
- Control of conception; and which does not achieve its principal intended action in or on the human body by pharmacological, immunological, or metabolic means, but which may be assisted in its function by such means
▶ Medical Device Classification
MFDS is classifying medical devices based on its potential impact on human like the following
| Class I: No potential risk |
※ Criteria for potential risk
|
| Class II: Low potential risk | |
| Class III: Medium potential risk | |
| Class IV: High potential risk |
| Class I | Class II, III, IV (No clinical needed) |
Class II, III, IV (Clinical needed) |
|
|---|---|---|---|
| Manufacturing | Product Register | Product Approval | Product Approval (Clinical) |
| Importing | Product Register | Product Approval | Product Approval (Clinical) |
▶ Manufacturing/Importing Business / Product registration and Approval
▶ Conformance acceptance categori
- Initial inspection: Initial inspection for newly registered product and manufacture
- Regular inspection: Every 3 years after initial inspection (should apply 90 days before expiration)
- Additional inspection: Adding new product category in scope
- Change inspection: Manufacturing address changes (except small storage area and laboratory which is not related with product quality)