


We have created a awesome theme
Far far away,behind the word mountains, far from the countries
 Medical Device Consulting > International
Medical Device Consulting > International
Medical Device Consulting
International Consulting
FDA
US FDA : Federal Food, Drug & Cosmetic Act
▶ FDA?
FDA's Center for Devices and Radiological Health (CDRH) is responsible for regulating firms who manufacture, repackage, relabel, and/or import medical devices sold in the United States. In addition, CDRH regulates radiation-emitting electronic products (medical and non-medical) such as lasers, x-ray systems, ultrasound equipment, microwave ovens and color televisions
The basic regulatory requirements that manufacturers of medical devices distributed in the U.S. must comply with are:
• Establishment registration,
• Medical Device Listing,
• Premarket Notification 510(k), unless exempt, or Premarket Approval (PMA),
• Investigational Device Exemption (IDE) for clinical studies
• Quality System (QS) regulation,
• Labeling requirements, and
• Medical Device Reporting (MDR)
▶ Relevant Standards and Regulations
- FD&C Act (Federal Food, Drug & Cosmetic Act)
- 21 CFR (The Code of Federal Regulations)
▶ Applicable Products
Medical devices are classified into Class I, II, and III. Regulatory control increases from Class I to Class III. The device classification regulation defines the regulatory requirements for a general device type. Most Class I devices are exempt from Premarket Notification 510(k); most Class II devices require Premarket Notification 510(k); and most Class III devices require Premarket Approval. A description of device classification and a link to the Product Classification Database is available at "Classification of Medical Devices."
▶ Classifications
A medical device is classified in one of three different classes. The classification identifies the level of regulatory control that is necessary to assure the safety and effectiveness of a medical device. Unless exempt, it also determines the marketing process (either 510(k) or PMA) the manufacturer must complete in order to obtain FDA clearance/approval for marketing.
Products are grouped in classification panels and get assigned a specific product code based on the classification designated under 21 CFR 862-892.
| Class I (low risk) | - These devices need "general controls". Compliance with the following items is necessary: Registration of device manufacturers and distributors, record keeping requirements, labeling requirements and a quality system that complies with the QSR requirements (including complaint files). |
|---|---|
| Class II (intermediate risk) | - "General controls" and "Special Controls" are required for Class II devices to provide the assurance of safety and efficacy.
- "Special Controls" include compliance with performance standards, post market surveillance schemes to track product defects and adherence to various guidelines and recommendations. - Required is a 510(k) unless there is a device exemption. |
| Class III (high risk) | - These devices require the controls already detailed for Class I and II devices. Additional controls are required depending on the particular product code. Pre-market approval of the safety and efficacy of a Class III device is generally required before commercial distribution is allowed. |
▶ General Registration Process
The following requirements must be met before a device can be submitted to the FDA for clearance:
- An US designated agent must exist. For Siemens Healthineers business units located outside of the U.S.,
- Registration: The local representative is required to register the facilities and the medical devices. The registration must be renewed annually.
- Quality System Requirements (QSR 21 CFR 820): Manufacturers of finished devices must comply with the QSR regulations.
- Inspection and access to records. FDA has the authority to examine any facility or records associated with the manufacture, process, packs, transport or storage of medical devices.
Pre-market notification 510(k):
Establishes substantial equivalence to a predicate device. There are 3 different types of 510(k):
- Traditional 510(k)
- Special 510(k)
- Abbreviated 510(k)
The manufacturer decides which of the three 510(k) approaches is most suitable to the product.
Note: 510(k) submission can be done via the designated agent or via Third Party Review, in case the device is eligible for Third Party Review. FDA maintains a list of devices that are eligible for Third Party Review at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfThirdParty/current.cfm.
Pre-market approval (PMA)
A PMA is required for Class III products and where it is not possible to demonstrate equivalence with an already commercially distributed device. E.g. a new clinical application for an existing device or a new device has been developed.

CE
CE Marking (EU)
▶ CE Marking?
CE Marking is unified registration approval mark in EU region which is saying all requirements for safety, health, environment and customer protection per EU policy. For distributing products in EU region, CE marking should be attached in product, package and accompanying documents. CE mark is guaranteeing to be distributed in EU region without any restriction.
▶ Relevant Standards and Regulations
- EN ISO 13485
- Council Directive 93/42/EEC of 14 June 1993 concerning medical devices
▶ Applicable products
CE / MDD(93/42/EEC) General Medical Device, CE / IVD(98/79/EC) IVD, CE / AIMDD(93/68/EEC) AIMDD
▶ Classifications
Medical Devices are classified into the following four classes according to the Medical Device Directive (MDD) Article 9: Class I, Class I sterile or with measuring function, Class IIa, Class IIb and Class III medical devices. The classification rules are defined in Annex IX of the MDD. Practical guidance and illustrative examples can be found in the MEDDEV 2.4/1 Classification of medical devices and in the Manual on borderline and classification in the Community Regulatory framework for medical device
※ Classification Criteria
Duration: Temporary-60min, short term-less than or equal to 30 days, long term-more than 30 days
Invasive device: Inserted into body as a whole or part through body holes or skin
Active device: Devices using electrical energy or power
※ Classification
Non-invasive devices : Rule 1∼4
Invasive devices : Rule 5∼8
Active device : Rule 9∼12
Special rules : Rule 13∼18
▶ General Registration Process
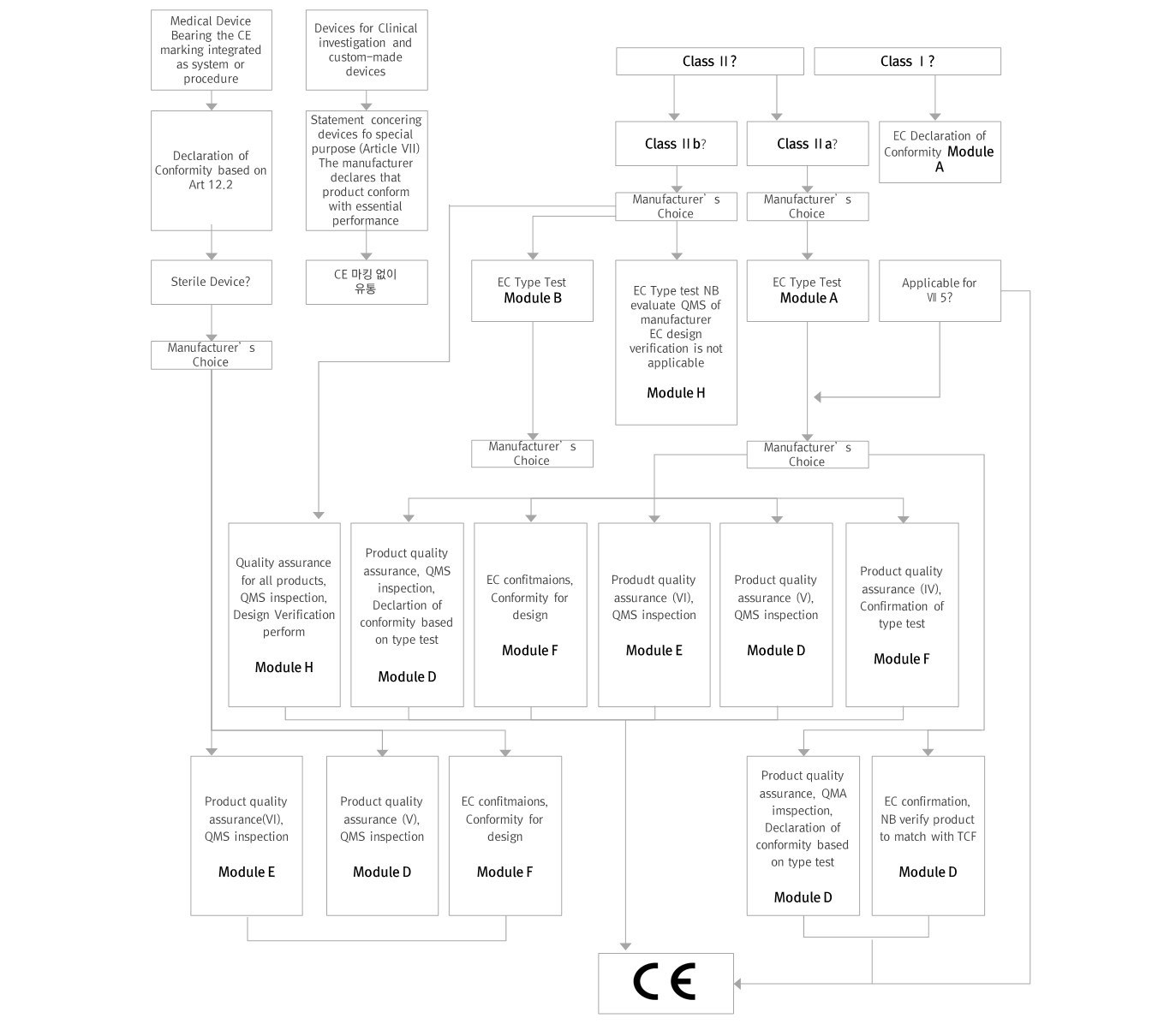
TGA
TGA (Australia)
▶ TGA?
The Therapeutic Goods Administration (TGA) is the regulatory body for therapeutic goods (including medicines, medical devices, gene technology, and blood products) in Australia. It is a Division of the Australian Department of Health established under the Therapeutic Goods Act 1989 (Cth).The TGA is responsible for conducting assessment and monitoring activities to ensure that therapeutic goods available in Australia are of an acceptable standard and that access to therapeutic advances is in a timely manner. Applicant should reside in Australia and CE is accepted in Australia. However some specific device might go through mandatory inspection from Australia in that case as well.
▶ Relevant Standards and Regulations
Therapeutic Goods Act 1989
Therapeutic Goods (Medical devices) regulations 2002
▶ Applicable products
The Act defines a medical device as:
a) Any instrument, apparatus, appliance, material or other article (whether sued alone or in combination, and including the software necessary for its proper application)) intended, by the person under whose name it is or is to be supplied, to be used for human beings for the purpose of one or more of the following:
i. Diagnosis, prevention, monitoring, treatment or alleviation of disease;
ii. Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap;
iii. Investigation, replacement or modification of the anatomy or of a physiological process; controls of conception;
and that does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but that may be assisted in its function by such means; or
b) An accessory to such an instrument, apparatus, appliance, material or other article.
▶ Classifications
The regulatory framework has a classification system for medical devices. Risk-based classification rules (Schedule 2 of the Regulations) must be applied. The classifications and levels of risk are:
| Classification | Level of Risk |
|---|---|
| Class I | Low |
| Class I – supplied sterile Class I – incorporating a measuring function Class IIa | Low-medium |
| Class Ⅱb | Medium-high |
| Class Ⅲ | High |
| Active implantable medical device (AIMD) | High |
The manufacturer is responsible for determining the classification of a device using a set of classification rules based on the:
• manufacturer’s intended use of the device
• level of risk to patients, users and other persons (the probability of occurrence of harm and the severity of that harm)
• degree of invasiveness in the human body
• duration of use. Identical devices may be classified differently if they are to be used in different parts of the body.
This is why the manufacturer’s intended use of the device is so critical to determining the appropriate classification
▶ General Registration Process
1. Manufacturer Approval
- Manufacturer’s evidence
1) Manufacturer should be registered in ARTG (Australian Register of Therapeutic Goods), it can be checked in TGA website and application can be go through TGA e-business (eBS)
2) Foreign manufacturer should have local sponsor in Australia for registering in ARTG. Sponsor should be legal personnel or organization in Australia and has responsibility for submission of all required documents, reporting of post-production failure and pat ARTG fee as well
2. Product Approval
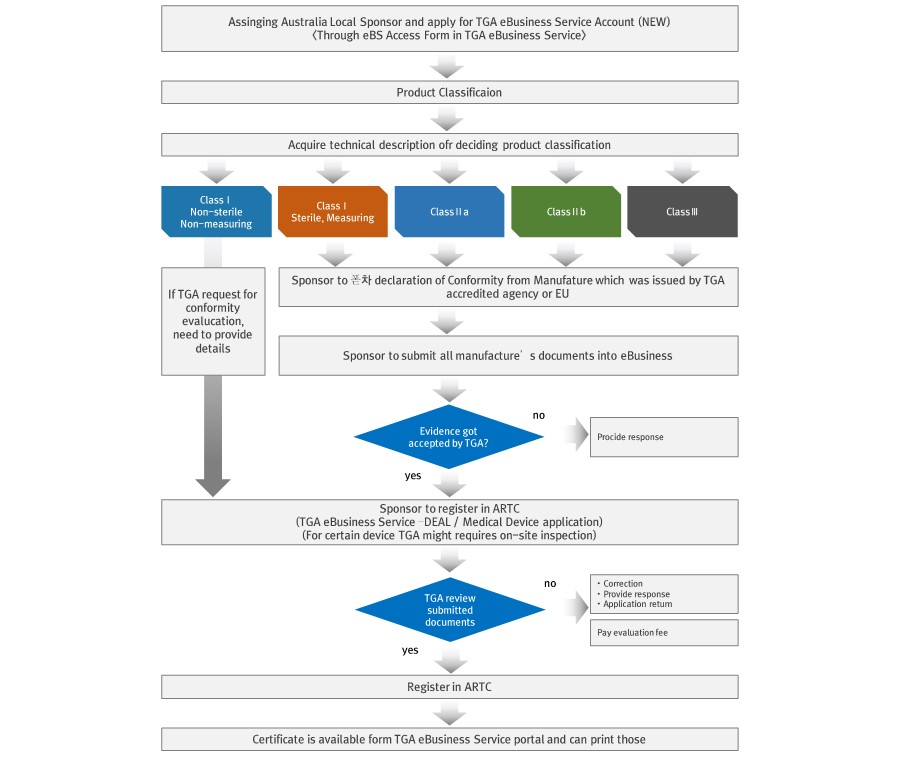
3. Quality Management System
ISO 13485
TGA does not require ISO 13485 certification separately since it is regarded as confirmed when product conformity evaluation.
Part1: Overall Quality assurance process
Part2: Type test (Sample test)
Part3: Verification process (Shipping test included)
Part4: Manufacturing quality assurance (Final inspection included)
Part5: Product Quality management system
Part6: Declaration of Conformity (Preparing technical documentation and PMS)
Part7: Special purpose medical device conformity evaluation process
Part8: Clinical evaluation process
ANVISA
ANVISA (Brazil)
Agencia Nacional de Vigilancia Sanitar
▶ ANVISA?
National Health Surveillance Agency (ANVISA) is the Brazilian authority which regulates Medical Devices. All Medical Devices have to be registered / filed (depending on the risk class) with National ANVISA before they are sold in Brazil.
ANVISA conducts biennial audits of Medical Device manufacturers selling in Brazil. The obtained ANVISA GMP certificate is valid for two years and can be extended for another 2 years by submission of a report by completion of a template provided by ANVISA
▶ Relevant Standards and Regulations
SINMETRO, ABNT, INMETRO, CONMETRO
▶ Applicable products
Drugs, Medical Device (including mechanical equipment), Foods, Cosmetics, and Detergents, Tabaco, Blood and blood related products are under ANVISA. ANVISA is applicable for local manufacturer, importers, distributors and hospitals. Definition of Medical device from ANVISA is like the followings;
Healthcare product, such as equipment, devices, materials, articles, or systems for medical, odonatological, or laboratory use or application, intended for prevention, diagnosis, treatment, rehabilitation, or anti-conception and that does not use pharmacological, immunological, or metabolic means to fulfill its main function in human beings, but can have its functions assisted by such means
▶ Classifications
Medical devices are divided into four different classes based on their potential risk to patient health or to the medical device operator.
- Class I Low risk – Requires simplified ANVISA registration
- Class II Medium risk – Required normal ANVISA registration
- Class III High risk – Required normal ANVISA registration
- Class IV High risk – Required normal ANVISA registration
A system consisting of different integrated or connected Medical Devices and/or accessories, which may each belong to different classes, will be classified in the most critical class. If a manufacturer is unsure to determine the classification of a Medical Device, a submission can be made to ANVISA who makes the final decision about the classification. If a Medical Device is intended to be used together with another device, classification rules will apply to each Medical Device. Accessories will be classified for themselves, separately from the Medical Device with which they are used. When more than one rule could apply the highest rule takes precedence.
▶ General Registration Process
[Brazil Medical Device Registration Flow chart]
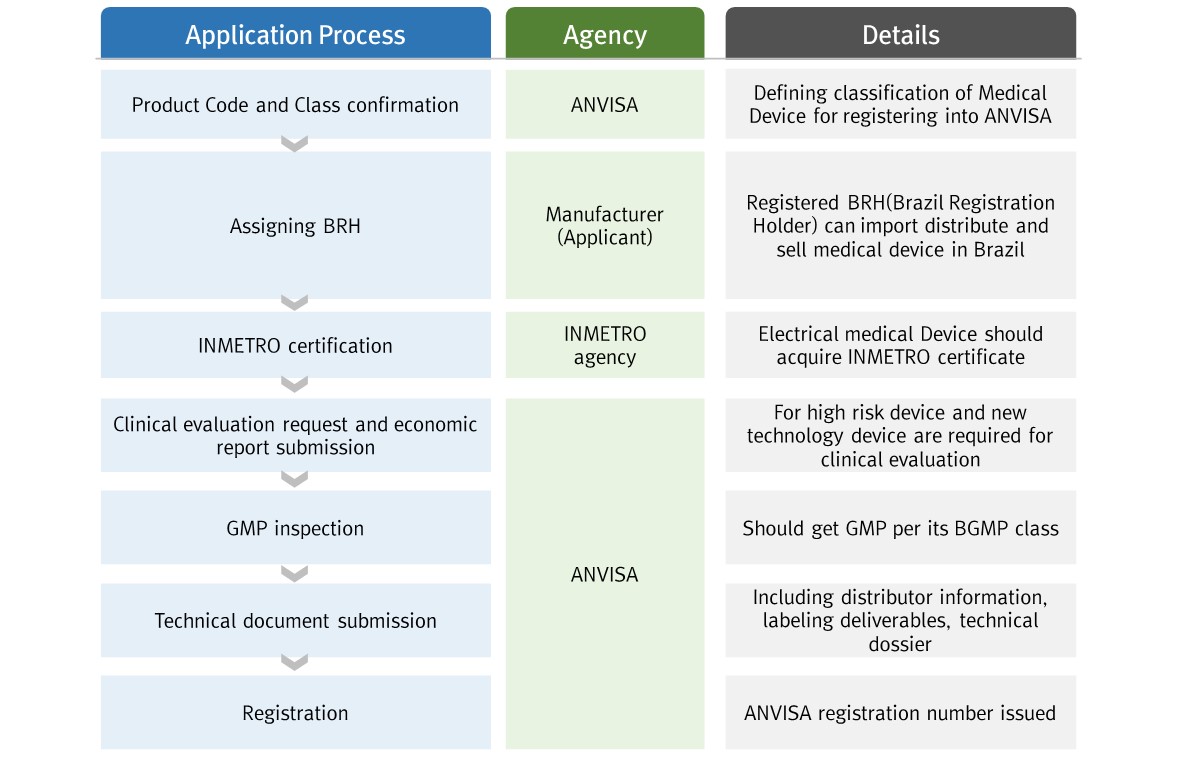
[ANVISA registration process]
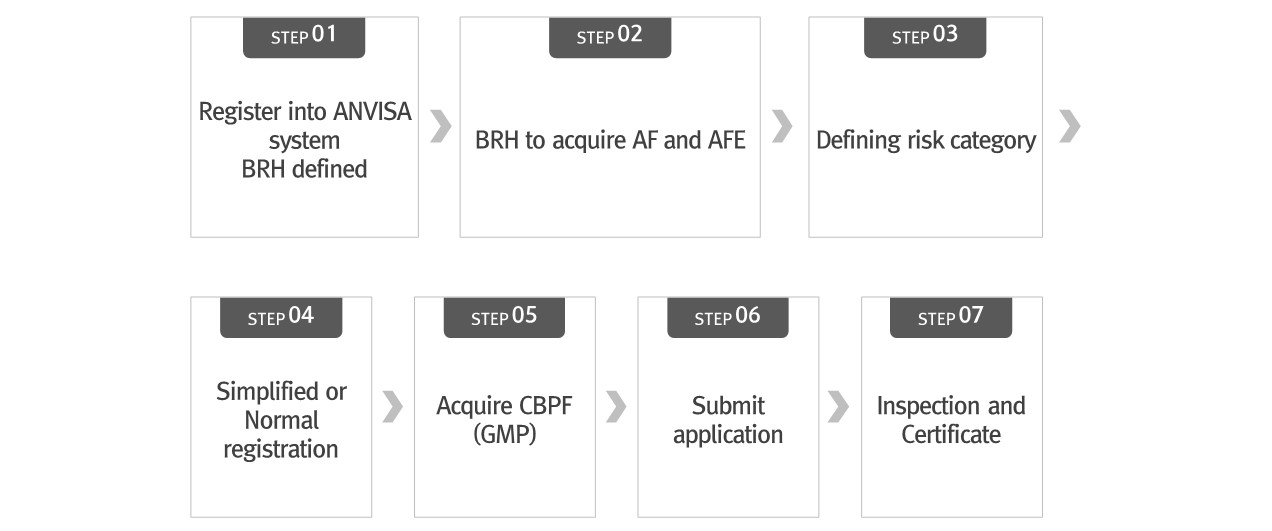
※Step 4, 5 can be proceeded simultaneously
* BRH: Brazil Registration Holder
* AF: Álvara de Funcionamento – Sales License
* AFE: Autorização de Funcionamento da Empresa – Sales permission
* GMP: Good Manufacturing Practice
* CBPF: Certificado de boas práticas de fabricação – Certificate for best manufacturer
Health Canada
Health Canada (Canada)
▶ Health Canada?
Device manufacturers must obtain a Medical Device License for Class II, III or IV medical devices. Health Canada’s Medical Device Bureau (MDB) grants the licenses. Sale of unlicensed devices is prohibited. However, offer for sale as part of an RFP of an unlicensed device is authorized, as long as the final sale of the device does not take place before the device has been authorized by Health Canada (as per Health Canada’s Announcement from 2013-09-04)
▶ Relevant Standards and Regulations
Food and Drug Act)
Canadian Medical Devices Regulation (CMDR, SOR/98-282)
▶ Applicable products
The manufacture and sale of medical devices are subject to the Food and Drugs Act and the Medical Devices Regulations. According to the Food and Drugs Act, a medical device is defined as "any article, instrument, apparatus or contrivance, including any component, part or accessory thereof, manufactured, sold or represented for use in human beings and animals for:
- the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state, or its symptoms,
- restoring, correcting or modifying a body function or the body structure,
- diagnosis of pregnancy, or
- care during pregnancy and at, and after birth of the offspring, including care of the offspring, and includes a contraceptive device but does not include a drug."
▶ Classifications
| Class | Risk level | Details |
|---|---|---|
| Class I | Lowest | Failure can cause very low impact to human body i.e. Bandage, toothbrush, hospital bed |
| Class Ⅱ | Low | Failure can cause low impact to human body i.e. Condom, tampon, surgical gloves, magnetic resonance imaging equipment |
| Class Ⅲ | Moderate | Failure can cause relatively high impact to human body i.e. Home glucose test kit, hip replacement implant, ultrasound diagnostic imaging equipment |
| Class Ⅳ | High | Impact is directly related with lives i.e. HIV test kit, implantable defibrillator, pacemaker |
▶ General Registration Process
Establishment license:
Importers and distributors of Medical devices (Class I to Class IV) must hold an establishment license. An existing license continues to be valid as long as a completed Annual Review application is received by Health Canada before April 1st with renewal fees, and comprehensive list of manufacturers whose devices are imported by Siemens in Canada
Medical Device License:
Class I devices do not need a license.
Class I devices do not need a license.
Medical Device Licenses are required for a Class II, III or Class IV product. They must be renewed annually. The degree of information which must be submitted to the MDB to obtain a license depends on the class of the device.
Quality System certificates:
Health Canada requires medical device manufacturers to use a quality system certificate as evidence of compliance to the appropriate regulatory quality system. Manufacturers of class II to IV devices must provide evidence that the devices which shall be sold in Canada are developed and manufactured in accordance with a certified quality system and are required to submit with their license application a copy of a valid Quality System certificate.
Note: Class I devices are exempt from device licensing and therefore exempt from quality system requirements.
For a two year period, starting January 1, 2017, Health Canada accepts certificates issued under both CMDCAS and MDSAP. According to Health Canada's MDSAP transition plan, CMDCAS certificates will no longer be accepted after December 31, 2018. Manufacturers will be required to submit valid MDSAP certificates by no later than January 1, 2019 in order to maintain their medical device licenses.
CFDA
CFDA : China Food & Drug Administration
▶ CFDA?
CFDA is responsible for protecting the public health by assuring the safety, efficacy, and security of drugs, biological products, Medical Devices, food supply and cosmetics. It is part of the Ministry of Health and has regulatory and legal enforcement functions. The China Food & Drug Administration (CFDA) is the only agency responsible for the product registration of imported medical devices. CMDE that is performing the technical review of the submission documents is a department below CFDA.
▶ Relevant Standards and Regulations
The State Council Decree No. 680/2016
The "Provisions for Medical Device Registration" (CFDA Order No. 4/2014 “Provisions for Medical Device Registration”
▶ Applicable products
The definition of Medical Devices is laid down in Article 76 of Order No. 680/2016 [1]: “Medical device” is an instrument, equipment, appliance, in vitro diagnostic reagent and its calibrator, material, and other similar or relevant articles including necessary computer software, directly or indirectly contacting human body; the effectiveness are obtained mainly through physical means other than pharmacological, immunological or metabolic ways, or such ways are involved in but only play auxiliary roles; which is used to achieve the following intended outcomes:
I. Diagnosis, prevention, monitoring, treatment or alleviation of diseases
II. Diagnosis, monitoring, treatment, alleviation or functional compensation for an injury
III. Examination, substitution, regulation or support of physiological structure or physiological process
IV. Life support or life sustaining
V. Contraception Control;
VI. Examination of the sample from human body to provide information for medical or diagnostic purpose.
The manufacturer has to compare the definition with the intended use and determine if the product is a Medical Device.
▶ Classifications
According to Order No. 680/2016 [1], Article 4 Medical Devices shall be classified and administrated based on risk level. There are three risk-based classes defined for medical devices:
Class I: are those with lower risk for which safety and effectiveness can be ensured through routine administration (e.g. film processing system, x-ray film, x-ray reading device);
Class II: are those with medium risk for which strict control is required to ensure their safety and effectiveness (e.g. x-ray tube housing assembly, Image intensifier);
Class III: are those with higher risk which special measures shall be taken and strict control is required to ensure their safety and effectiveness (e.g. MR, CT, X-ray, Nuclear Medicine)
▶ General Registration Process
Registration application for Medical Devices manufactured outside of P. R. China shall be reviewed and approved by CFDA. Approval time may range from minimum 5 months to around one year after submitting the submission documents, but it can differ depending on the submission of feedback in case of supplemental questions of CFDA. All documents that have to be submitted to CFDA for registration of Medical Devices are listed in Annex 1 GD 13 China Guidance “Medical Device Regulatory Requirements in P.R. China (Foreign Establishments)”
▶ Type Testing
Attention: Currently, the situation regarding type testing is still unclear. Order 680 is currently under rework of CFDA. According to the current draft it seems that type testing can be performed in manufacturer´s test lab according to applicable Chinese standards. This section will be updated as soon as reliable information is available.
Imported Medical Devices of Class II and Class III are subject to registration testing (type testing) by authorized Chinese test institutes using national standards. For extension registration type testing may be exempt if no Chinese mandatory standard was revised during validity period of the registration certificate. Type testing is normally performed by CFDA testing institutes engineers, either in Chinese test institutes or conducted at the manufacturer’s site for safety and performance testing as well as for EMC testing. For large scale medical devices (e.g. CT, MR, MI and some AX products) which have specific requirements about installation or which are difficult to test, type testing at manufacturer’s site is possible. Type testing is typically based on PTR, Operator Manual, Data sheets and other technical documents.
PMDA
PMDA : Pharmaceuticals and Medical Devices Agency
▶ PMDA?
All foreign establishments must be registered with PMDA through the MAH. MAH performs the registration on behalf of the Healthcare Business Area (HBAs) / Healthcare Business Lines (HBLs). The MAH completes the application form with
- general information (address, manufacturers name, management name, etc.) and
- a list of the manufactured products.
- Self declaration by manager of foreign manufacturing facilities.
- Personal history of responsible person of the facility
- Document to show the place of facility.
▶ Relevant Standards and Regulations
JPAL (Japan’s Pharmaceutical Affairs Law)
▶ Classifications and Applicable products
| Classification | Description | Examples | Potential risk |
|---|---|---|---|
| Class I | MD: General Medical Devices IVD: Analytes defined by MHLW | Diagnostic equipment, x-ray film, IVD instruments, Analytes (e.g. CRP, Mg, CSA, HbA1c etc.) | Almost insignificant (extremely low) |
| Class II | MD: Controlled Medical Devices IVD: Moderate Risk Class Analytes not Classified as Class I or III | Endoscopes, MRI, CT, ultrasound, tests for analytes (e.g. TACR, TnI, BNP, TSH, etc.) | Have potential risk (low) |
| Class III & IV | MD: Special controlled Medical Devices IVD: New and HighRisk Analytes (Class III only) | balloon catheters (III), stents (IV), valves (IV), linear accelerator (III), lithotriptor (III); tests for infectious diseases, cancer markers’ Antistreptolysin O, bacterial identification (e.g. HIV, HBs-Ag, CA19-9, HCV-Ab ) (III) | Significant (high or middle) |
▶ General Registration Process
|
Classification |
Submission category |
Regulatory clearance procedure |
|
|
Class I |
Notification |
No regulatory approval or marketing authorization is needed |
|
|
Class II |
If conformity assessment criteria applicable |
Certification |
RCB (3rd Party) certification is required. Only those class II devices for which PMDA has evaluation standards may utilize the RCB certification process. RCB reviews application and compliance with PMDA Essential Requirements and new QMS regulation. |
|
New technology |
Governmental approval (SHONIN) |
PMDA review of application and check of factory compliance to Essential Requirements and Good Manufacturing Practices through documentation or factory inspection required |
|
|
Class III (Designated by MHLW) |
Certification |
Class III medical device designated by MHLW require RCB certification. (Same to Class II certification) |
|
|
Class III & IV |
Governmental approval |
PMDA review of application and check of factory compliance to Essential Requirements and Good Manufacturing Practices through documentation or factory inspection required |
|
SFDA
SFDA
Saudi Food and Drug Authority
▶ SFDA ?
Healthcare products including establishments have to be registered before import or sale:
Enrolment is through the Saudi Food & Drug Authority’s (SFDA) internet site of the Medical Devices National Registry (MDNR).
The second step is the Medical Device Establishment Licensing System (MDEL) for medical devices establishments in Saudi Arabia. It is a web based system where all applications shall be applied online. To be able to apply for MDEL, the applicant has to be registered in MDNR
▶ Relevant Standards and Regulations
[ Laws ]
+ 2008-12 Medical Device Interim Regulation
+ 2010-01 Implementing rule on Designation and Oversight of Conformity AssessmentBodies (IR 1)
+ 2010-04 Implementing rule on Establishment Registration (IR 2)
+ 2010-04 Implementing Rule on Medical Devices Listing (IR 3)
+ 2011-02 Implementing rule on Establishment Licensing (IR 4)
+ 2011-02 Implementing Rule on Licensing of Authorized Representatives (IR5)
+ 2011 Implementing Rule on the Validation of Documents to be provided to the SFDA by Manufacturers for Marketing Authorization (IR 6)
+ 2010-01 Implementing Rule on Post-Marketing Surveillance (IR 7)
+ 2010-01 Implementing Rule on Safeguard Procedures (IR 8)
+ 2018-06 Updated Medical Device Interim Regulation
▶ Applicable products
In fulfilment of Medical Devices Interim Regulation in addition to Implementing Rule on the Validation of Documents to be provided to the SFDA by Manufacturers for Marketing Authorization (MDS-IR6), The SFDA lunched Medical Devices marketing Authorization System (MDMA).
MDMA is an electronic system aims to authorize medical devices after they comply with the Medical Devices Interim Regulation (MDIR) and in particular to the implementing rule MDS-IR6 for Medical Device Marketing Authorization (MDMA). The system allows local manufacturers and overseas manufacturers authorized representatives to apply electronically for medical devices marketing authorization which permits relevant medical devices to be placed on the market of the Kingdom of Saudi Arabia, when satisfied that the applicant has provided all the required information for market authorization.
All medical devices & IVDs intended to be marketed in Saudi Arabia should have a valid Medical Device Marketing Authorization (MDMA) as per the following enforcement dates:
| Class | Type | Enforcement Date |
|---|---|---|
| High Risk | In Vitro Diagnostic | Oct 1st 2012 |
| Medical Device | Dec 1st 2012 | |
| Medium Risk | In Vitro Diagnostic | Dec 1st 2012 |
| Medical Device | Dec 31st 2014 | |
| Low Risk | In Vitro Diagnostic | Jun 30th 2015 |
| Medical Device | Dec 31st 2015 |
To obtain marketing authorization, medical devices shall comply with the relevant regulatory requirements applicable in one or more of the jurisdictions of Australia, Canada, Japan, the USA and the EU/EFTA, and additionally with provisions specific to Saudi Arabia concerning labeling and conditions of supply and/or use (Article 6, Medical Device Interim Regulation). Documents should be in English unless the SFDA has reached a prior agreement with the applicant that another language is acceptable.
▶ Classifications
Classification is based on GHTF founding member jurisdiction.
Additionally in case classification of product is not available, SFDA Medical Device Sector announced the launch of Medical Device Classification (MDC) system.
MDC SYSTEM is an online classification system to help the applicant in classification of the product to classify any product whether it is medical device or not and to determine whether the product fall in medical device responsibilities or not.
Starting May 1st, 2018, a new market pathway for Class I low-risk, non-sterile, non-measuring devices will become effective. Going forward there will be 2 market pathway options:
- Option 1: Obtain Market Authorization (standard Medical Device Market Application (MDMA)) for low-Risk, non-sterile and non-measure medical devices. Then, list these devices by Importers and distributors at the MDNR system.
- Option 2: List low risk (non-sterile/non-measure) medical devices by Importers and distributors through MDNR.
▶ General Registration Process
The SFDA issued a Medical Devices Interim Regulation (MDIR) on 2008-12-27. It specifies the overall framework of the regulatory approach for the Saudi marketing authorization and the post-marketing surveillance of medical devices. It applies to manufacturers, authorized representatives, importers and distributors and includes all medical devices and their accessories that will be supplied to Saudi Arabia.
Eight Implementing Rules (IR) have been adopted by the SFDA to specify and complete the general provisions of the MDIR. They have force of law. Their application takes part in ensuring that medical devices placed on the Saudi Arabian market achieve an appropriate level of safety and performance with regard to their manufacture, supply and use. These are:
8 rules
+ IR #1 Designation and oversight of conformity assessment bodies
+ IR #2 Establishment registration
+ IR #3 Medical devices listing
+ IR #4 Establishment licensing
+ IR #5 Licensing of authorized representatives
+ IR #6 Validation of documents to be provided to the SFDA by manufacturers for marketing authorization
+ IR #7 Post-marketing surveillance
+ IR #8 Safeguard procedures
MoHW
MoHW (Taiwan)
▶ MoHW?
The Ministry of Health and Welfare (MoHW) is the local health authority that regulates the import of (in-vivo) Medical Devices in Taiwan. If a foreign manufacturer wants to market a medical device in Taiwan,
- Quality System Documentation (QSD) approval and
- Product pre-market registration
By the Bureau Taiwan Food and Drug Administration (TFDA) is necessary before an import license for the affected device will be issued to Siemens Healthcare Limited (SHL). The fulfillment of these requirements is a prerequisite for a foreign medical device manufacturer to import its devices to Taiwan. Only if TFDA issues an import license to SHL the product can be imported.
▶ Relevant Standards and Regulations
Pharmaceutical Affairs Act
Regulations Governing Management of Medical Devices
▶ Applicable products
MoHW has established a classification system for different generic types of devices and grouped them into 17 medical categories. Each device is assigned to a Class from I to III by degree of risk (low risk to high risk) to assure the safety and effectiveness of the device. The final classification of medical devices is specified by MoHW.
The categories of medical devices are defined in the Annex I of the Regulation “Governing Management of Medical Device”.
Medical devices are currently classified into 17 categories:
(1) Clinical Chemistry and Clinical Toxicology
(2) Hematology and Pathology
(3) Immunology and Microbiology
(4) Anesthesiology
(5) Cardiovascular Medical Science
(6) Dentistry
(7) Ear Nose and Throat
(8) Gastroenterology and Urology
(9) General and Plastic Surgery
(10) General Purpose Apparatus for Hospital and Individual Use
(11) Neuroscience
(12) Obstetrics and Gynecology
(13) Ophthalmology
(14) Orthopedics
(15) Physical Therapy
(16) Radiology
(17) Others as determined by the central competent health authority
▶ Classifications and General Registration Process
The necessary requirements for registration of medical devices are specified in the "Regulations Governing Management of Medical Devices" which is based on the classification of the devices
|
Classification |
QSD |
Product Registration |
|
Class I device |
QSD requirements must be fulfilled by the manufacturer |
Pre-market product registration necessary |
|
Class II device |
QSD requirements must be fulfilled by the manufacturer. |
Pre-market product registration necessary |
|
Class III device |
QSD requirements must be fulfilled by the manufacturer. |
Pre-market product registration and Clinical trial necessary. |
The specified requirements and an application form for registration and market approval of medical device must be submitted to TFDA for pre-market registration. After successful registration of the medical device the QSD approval letter and the pre-market registration notification will be released by TFDA. The pre-market product registration can take significantly longer than 6 and 8 months due to the new two steps review procedure.
Russia
Roszdravnadzor (Russia)
▶ Roszdravnadzor?
All medical devices and IVD products must be registered by the Federal Service of Health Care and Social Development Control (ROSZDRAVNADZOR). The main objective of the registration is approval for importation, sales and application in Russia.
Registration shall be performed based on the results of the technical and clinical tests and appraisals evidencing quality, efficiency and safety of products. The appraisals are done on a phase-by-phase basis:
− the first phase - expert check of the registration dossier to define possibility (impossibility) of MD clinical tests performance;
− the second phase - expert check of completeness and results of performed technical, toxicological, clinical tests and final decision on registering the medical devices the technical, clinical, toxicological tests are performed by accredited independent investigation centers
▶ Relevant Standards and Regulations
- Federal Law of N 323 FZ: "On the fundamentals of health protection in the Russian Federation"
- Decree N1416 "Rules of state registration of medical devices" (2012.12.27.)
▶ Applicable products
Any medical appliances, apparatuses, devices, equipment, materials, and other products used for medical purposes either separately or in combination with each other and with other accessories required for the use of these products as intended, including customized software, and designed manufacturer (producer) for the prevention, diagnosis, treatment and aftercare of diseases, monitoring of the human body for medical research, medical tests, rehabilitation, replacement, modification of anatomy or physiological functions of the body, pregnancy prevention or termination, the functional purpose of which is not implemented by pharmacological, immunological, genetic or metabolic impact on the human body (hereinafter “medical devices”) Government Decree of 27/12/2012 No. 1416, Art. 2
▶ Classifications
The MoH has implemented a medical device classification system identical to the one in the European Communities.
Classification shall be made depending the degree of potential risk of use for medical purposes; each product shall fall to anyone of the following four classes:
· Class 3 – medical products with high risk level;
· Class 2b – medical products with increased risk level;
· Class 2a – medical products with medium risk level;
· Class 1 – medical products with low risk level.
Medical products being diagnostic kits (in vitro) shall be classified as follows:
· Class 3 medical products with high individual risk and/or high public health risk
· Class 2b - medical products with high individual risk and/or moderate public health risk;
· Class 2a - medical products with moderate individual risk and/or low public health risk;
· Class 1 - medical products with low individual risk and low public health risk.
▶ General Registration Process
Licensing
Imported medical devices are subject to certification – to ensure conformity with the relevant safety requirements and local standards.
Successful product registration is precondition for certification. For imported medical device (except IVD) which has to fulfill the requirements regarding the relevant standards a Declaration of Conformity must be obtained. The declaration of conformity will be issued to the name of the importing organization that has to be a Russian legal entity. These documents can be issued by one of the accredited bodies in Russia.
For IVD products voluntary Certificate of Conformity can be obtained for sales support (for tenders, etc). Voluntary Certificate of Conformity, as well as Declaration of Conformity must be extended annually. Renewal of certificates issued after 2005 are handled by the regional unit located in Moscow. Documents to be submitted vary by product and by certification status (new/renewal). Typical information requested:
− Local Test reports on IEC standards
− ISO certificates (9001, 13485)
− Copy of EC Declaration of Conformity (DoC).
− Copy of EC Certificate
− Valid registration certificate
− Operating Manual of the medical device in country language
− Agreement (on the quality guarantees and non-compliance of the products to the quality requirements, please see example below).
Local Technical Testing is a part of certification / re-certification process. Typically test reports that were done for registration purpose can be used.
Average time frame to obtain the certificate is 1 month.
Registration procedures of medical devices and IVD products are regulated by DECISION OF THE GOVERNMENT OF THE RUSSIAN FEDERATION NO. 1416 OF DECEMBER 27, 2012 ON THE APPROVAL OF RULES FOR THE STATE REGISTRATION OF MEDICAL ARTICLES.
Registration approval timeframe is 50 working days after submission of the registration dossier. The period of clinical trials of a medical product shall not be included in this 50-days period. The period of validity of Registration certificates issued after January 1, 2013 is unlimited. The Registration certificates issued before January 1, 2013 with a defined expiration date are valid until expiry of the certificate.
All Registration certificates issued before January 1, 2013 shall be replaced before January 1, 2017.
The replacement of the registration certificate is performed via filing at the ROSZDRAVNADZOR
List of the documents required for registration:
− Inventory/summary document of the documents prepared for submission Application letter for registration of a medical product
− Confirmation document about the payment of the government registration fee
− Applicant company’s business licence
− Normative document (list of applicable standards)
− Certificate for measuring device (applicable for medical device classified as measuring device in the metrology regulation)
− Photos of all product parts and accessories
− Legalized copy with apostil stamp of: EC Declaration of Conformity, EC Certificate
− Legalized copy with apostil stamp of ISO 9001 and/ ISO 13485 Certificates
− Operating Manual of the medical device in country language
− Data Sheet
− Risk Management File
− Test reports (for safety and performance, biocompatibility and EMC).
− Technical file extract: Documents regulating the structure of medical device, determining technical requirements and including the information for their development, manufacture, application, operation, maintenance, repair, and destruction
− Documents containing data on the medical device clinical use, reviews, scientific reports, publications, presentations, risk analysis, medical device application methods in particular (if any)
− Documents specifying the composition of materials, contacting the human body surface Reports of toxicological studies (biocompatibility tests) (if any).
− Legalized copy with apostil stamp of Power of attorney authorizing the local representative to act on behalf of the manufacturer (see Power of Attorney_Template)
− Legalized copy with apostil stamp of Establishment registration document for legal manufacturer and manufacturing sites (incl. OEM) e.g. extract from the United States register of juridical persons (EGRUL) or extract from the register of the company by the authorized authority in the country of the registration of the medical device manufacturer (e.g. “Handelsregisterauszug”).
All documents submitted to the authority must be in Russian language.